本文简要介绍 python 语言中 scipy.stats.spearmanr 的用法。
用法:
scipy.stats.spearmanr(a, b=None, axis=0, nan_policy='propagate', alternative='two-sided')#计算 Spearman 相关系数和相关 p 值。
Spearman rank-order 相关系数是两个数据集之间关系单调性的非参数度量。与其他相关系数一样,该相关系数在 -1 和 +1 之间变化,其中 0 表示不相关。 -1 或 +1 的相关性意味着精确的单调关系。正相关意味着随着 x 的增加,y 也会增加。负相关意味着随着 x 的增加,y 会减少。
p 值粗略地表示不相关系统生成的数据集的 Spearman 相关性至少与根据这些数据集计算出的相关性一样极端的概率。尽管 p 值的计算并未对样本背后的分布做出强有力的假设,但它仅对于非常大的样本(> 500 个观测值)才准确。对于较小的样本量,请考虑排列测试(请参阅下面的示例部分)。
- a, b: 1D 或 2D 数组,b 是可选的
一个或两个包含多个变量和观测值的一维或二维数组。当这些是一维时,每个代表单个变量的观察向量。对于 2-D 情况下的行为,请参见下面的
axis。两个数组需要在axis维度中具有相同的长度。- axis: int 或无,可选
如果axis=0(默认),则每列代表一个变量,行中包含观察值。如果axis = 1,则关系被转置:每一行代表一个变量,而列包含观察值。如果axis=None,那么两个数组都将被解开。
- nan_policy: {‘propagate’, ‘raise’, ‘omit’},可选
定义当输入包含 nan 时如何处理。以下选项可用(默认为‘propagate’):
‘propagate’:返回 nan
‘raise’:引发错误
‘omit’:执行忽略 nan 值的计算
- alternative: {‘双面’,‘less’, ‘greater’},可选
定义备择假设。默认为“双面”。可以使用以下选项:
“双边”:相关性不为零
‘less’:相关性为负(小于零)
‘greater’:相关性为正(大于零)
- res: SignificanceResult
包含属性的对象:
- 统计 float 或 ndarray(二维正方形)
Spearman 相关矩阵或相关系数(如果仅给出 2 个变量作为参数)。相关矩阵是正方形,长度等于
a和b中变量(列或行)总数的总和。- p值 浮点数
假设检验的 p 值,其原假设是两个样本没有序数相关性。有关替代假设,请参阅上面的替代方案。 pvalue 与统计量具有相同的形状。
ConstantInputWarning如果输入是常量数组,则引发。这种情况下没有定义相关系数,所以返回
np.nan。
参数 ::
返回 ::
警告 ::
参考:
[1]Zwillinger, D. 和 Kokoska, S. (2000)。 CRC 标准概率和统计表和公式。查普曼和霍尔:纽约。 2000. 第 14.7 条
[2]肯德尔,M.G. 和斯图尔特,A. (1973)。高级统计理论,第 2 卷:推理和关系。格里芬。 1973.第31.18条
[3]Kershenobich, D.、Fierro, F. J. 和 Rojkind, M. (1970)。人肝硬化中游离脯氨酸池与胶原蛋白含量的关系。临床研究杂志,49(12), 2246-2249。
[4]Hollander, M.、Wolfe, D. A. 和 Chicken, E. (2013)。非参数统计方法。约翰·威利父子。
[5]B. Phipson 和 G. K. Smyth。 “排列 P 值不应该为零:随机抽取排列时计算精确的 P 值。”遗传学和分子生物学中的统计应用 9.1 (2010)。
[6]勒德布鲁克,J. 和达德利,H. (1998)。为什么排列检验在生物医学研究中优于 t 和 F 检验。 《美国统计学家》,52(2), 127-132。
例子:
考虑以下来自[3]的数据,该数据研究了不健康人类肝脏中游离脯氨酸(一种氨基酸)和总胶原蛋白(一种常见于结缔组织中的蛋白质)之间的关系。
下面的
x和y数组记录了两种化合物的测量值。观察结果是配对的:每个游离脯氨酸测量值均取自同一肝脏,作为相同index下的总胶原蛋白测量值。>>> import numpy as np >>> # total collagen (mg/g dry weight of liver) >>> x = np.array([7.1, 7.1, 7.2, 8.3, 9.4, 10.5, 11.4]) >>> # free proline (μ mole/g dry weight of liver) >>> y = np.array([2.8, 2.9, 2.8, 2.6, 3.5, 4.6, 5.0])在[4]中使用斯皮尔曼相关系数对这些数据进行了分析,这是一种对样本之间单调相关性敏感的统计量。
>>> from scipy import stats >>> res = stats.spearmanr(x, y) >>> res.statistic 0.7000000000000001对于具有强正序相关的样本,该统计量的值往往较高(接近 1);对于具有强负序相关的样本,该统计量的值往往较低(接近 -1);对于具有强负序相关的样本,该统计量的值往往较小(接近于 0)。具有弱序数相关性。
该检验是通过将统计观察值与零分布进行比较来进行的:零假设下得出的统计值分布,即总胶原蛋白和游离脯氨酸测量值是独立的。
对于此测试,可以转换统计量,使得大样本的零分布是具有
len(x) - 2自由度的 Student t 分布。>>> import matplotlib.pyplot as plt >>> dof = len(x)-2 # len(x) == len(y) >>> dist = stats.t(df=dof) >>> t_vals = np.linspace(-5, 5, 100) >>> pdf = dist.pdf(t_vals) >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> def plot(ax): # we'll re-use this ... ax.plot(t_vals, pdf) ... ax.set_title("Spearman's Rho Test Null Distribution") ... ax.set_xlabel("statistic") ... ax.set_ylabel("probability density") >>> plot(ax) >>> plt.show()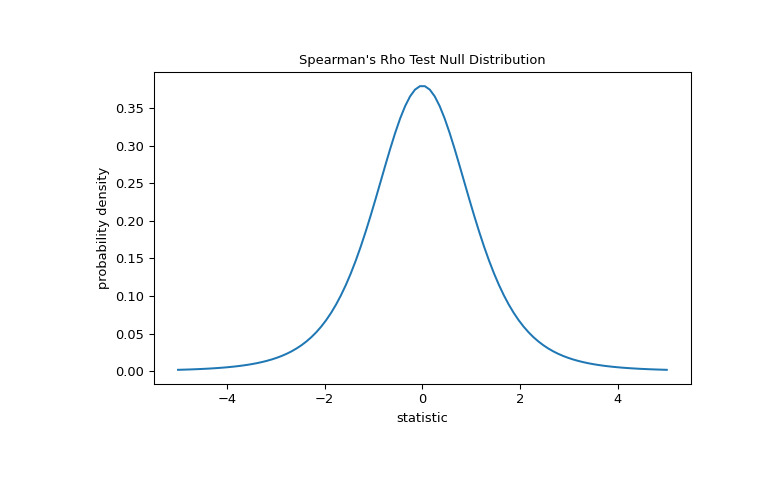
比较通过 p 值进行量化:零分布中比统计观察值极端或更极端的值的比例。在统计量为正的双边测试中,大于转换统计量的零分布元素和小于观察统计量负值的零分布元素均被视为 “more extreme”。
>>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> rs = res.statistic # original statistic >>> transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs))) >>> pvalue = dist.cdf(-transformed) + dist.sf(transformed) >>> annotation = (f'p-value={pvalue:.4f}\n(shaded area)') >>> props = dict(facecolor='black', width=1, headwidth=5, headlength=8) >>> _ = ax.annotate(annotation, (2.7, 0.025), (3, 0.03), arrowprops=props) >>> i = t_vals >= transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> i = t_vals <= -transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> ax.set_xlim(-5, 5) >>> ax.set_ylim(0, 0.1) >>> plt.show()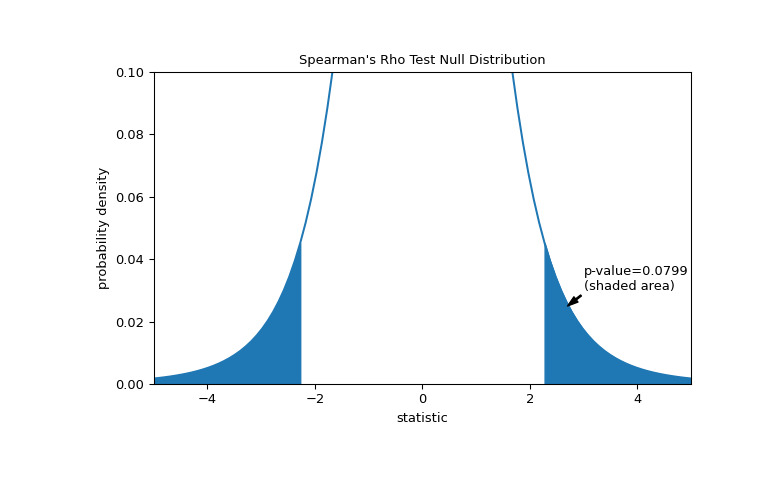
>>> res.pvalue 0.07991669030889909 # two-sided p-value如果 p 值为 “small” - 也就是说,如果从产生如此统计量极值的独立分布中采样数据的概率较低 - 这可以作为反对零假设的证据,支持替代方案:总胶原蛋白和游离脯氨酸的分布不是独立的。注意:
反之则不成立;也就是说,检验不用于为原假设提供证据。
将被视为 “small” 的值的阈值是在分析数据之前应做出的选择 [5],同时考虑误报(错误地拒绝原假设)和漏报(未能拒绝假设)的风险。错误的原假设)。
p 值小并不能证明效果大;相反,它们只能为 “significant” 效应提供证据,这意味着它们不太可能在原假设下发生。
假设在进行实验之前,作者有理由预测总胶原蛋白和游离脯氨酸测量值之间存在正相关性,并且他们选择评估零假设相对于片面替代方案的合理性:游离脯氨酸具有正相关性与总胶原蛋白的顺序相关。在这种情况下,只有零分布中与观察到的统计量一样大或更大的值才被认为是更极端的。
>>> res = stats.spearmanr(x, y, alternative='greater') >>> res.statistic 0.7000000000000001 # same statistic >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> pvalue = dist.sf(transformed) >>> annotation = (f'p-value={pvalue:.6f}\n(shaded area)') >>> props = dict(facecolor='black', width=1, headwidth=5, headlength=8) >>> _ = ax.annotate(annotation, (3, 0.018), (3.5, 0.03), arrowprops=props) >>> i = t_vals >= transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> ax.set_xlim(1, 5) >>> ax.set_ylim(0, 0.1) >>> plt.show()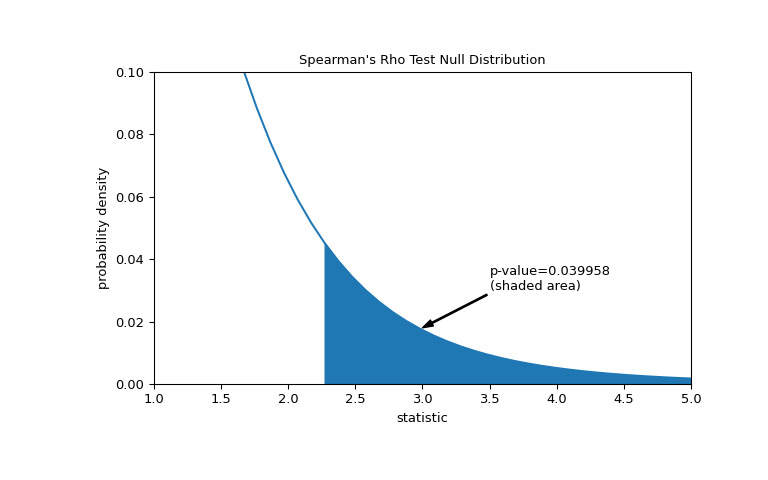
>>> res.pvalue 0.03995834515444954 # one-sided p-value; half of the two-sided p-value请注意,t 分布提供了零分布的渐近近似;它仅对于具有许多观测值的样本是准确的。对于小样本,进行排列检验可能更合适:在总胶原蛋白和游离脯氨酸独立的零假设下,每个游离脯氨酸测量值与任何总胶原蛋白测量值同样可能被观察到。因此,我们可以组建一个精确的通过计算之间每个可能的元素配对下的统计量来计算空分布
x和y.>>> def statistic(x): # explore all possible pairings by permuting `x` ... rs = stats.spearmanr(x, y).statistic # ignore pvalue ... transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs))) ... return transformed >>> ref = stats.permutation_test((x,), statistic, alternative='greater', ... permutation_type='pairings') >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> ax.hist(ref.null_distribution, np.linspace(-5, 5, 26), ... density=True) >>> ax.legend(['aymptotic approximation\n(many observations)', ... f'exact \n({len(ref.null_distribution)} permutations)']) >>> plt.show()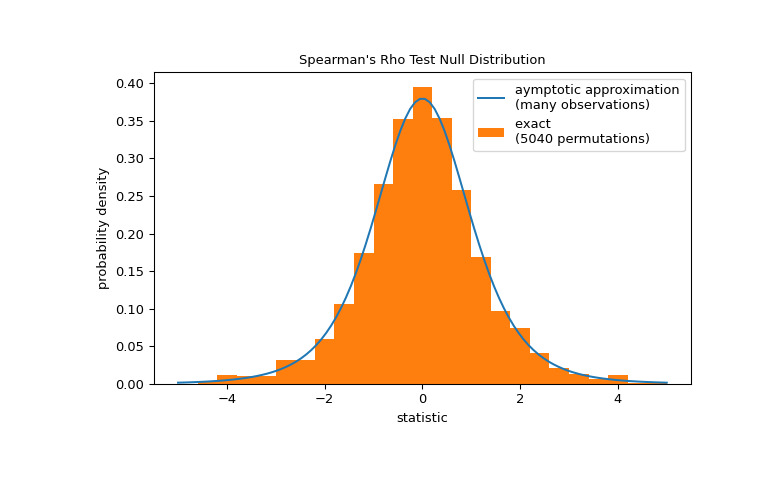
>>> ref.pvalue 0.04563492063492063 # exact one-sided p-value
相关用法
- Python SciPy stats.special_ortho_group用法及代码示例
- Python SciPy stats.skewnorm用法及代码示例
- Python SciPy stats.semicircular用法及代码示例
- Python SciPy stats.skellam用法及代码示例
- Python scipy.stats.somersd用法及代码示例
- Python SciPy stats.sem用法及代码示例
- Python SciPy stats.siegelslopes用法及代码示例
- Python SciPy stats.skewcauchy用法及代码示例
- Python SciPy stats.scoreatpercentile用法及代码示例
- Python SciPy stats.skew用法及代码示例
- Python SciPy stats.shapiro用法及代码示例
- Python SciPy stats.studentized_range用法及代码示例
- Python SciPy stats.sigmaclip用法及代码示例
- Python SciPy stats.skewtest用法及代码示例
- Python SciPy stats.sobol_indices用法及代码示例
- Python SciPy stats.anderson用法及代码示例
- Python SciPy stats.iqr用法及代码示例
- Python SciPy stats.genpareto用法及代码示例
- Python SciPy stats.cosine用法及代码示例
- Python SciPy stats.norminvgauss用法及代码示例
- Python SciPy stats.directional_stats用法及代码示例
- Python SciPy stats.invwishart用法及代码示例
- Python SciPy stats.bartlett用法及代码示例
- Python SciPy stats.levy_stable用法及代码示例
- Python SciPy stats.page_trend_test用法及代码示例
注:本文由纯净天空筛选整理自scipy.org大神的英文原创作品 scipy.stats.spearmanr。非经特殊声明,原始代码版权归原作者所有,本译文未经允许或授权,请勿转载或复制。