本文簡要介紹 python 語言中 scipy.stats.spearmanr 的用法。
用法:
scipy.stats.spearmanr(a, b=None, axis=0, nan_policy='propagate', alternative='two-sided')#計算 Spearman 相關係數和相關 p 值。
Spearman rank-order 相關係數是兩個數據集之間關係單調性的非參數度量。與其他相關係數一樣,該相關係數在 -1 和 +1 之間變化,其中 0 表示不相關。 -1 或 +1 的相關性意味著精確的單調關係。正相關意味著隨著 x 的增加,y 也會增加。負相關意味著隨著 x 的增加,y 會減少。
p 值粗略地表示不相關係統生成的數據集的 Spearman 相關性至少與根據這些數據集計算出的相關性一樣極端的概率。盡管 p 值的計算並未對樣本背後的分布做出強有力的假設,但它僅對於非常大的樣本(> 500 個觀測值)才準確。對於較小的樣本量,請考慮排列測試(請參閱下麵的示例部分)。
- a, b: 1D 或 2D 數組,b 是可選的
一個或兩個包含多個變量和觀測值的一維或二維數組。當這些是一維時,每個代表單個變量的觀察向量。對於 2-D 情況下的行為,請參見下麵的
axis。兩個數組需要在axis維度中具有相同的長度。- axis: int 或無,可選
如果axis=0(默認),則每列代表一個變量,行中包含觀察值。如果axis = 1,則關係被轉置:每一行代表一個變量,而列包含觀察值。如果axis=None,那麽兩個數組都將被解開。
- nan_policy: {‘propagate’, ‘raise’, ‘omit’},可選
定義當輸入包含 nan 時如何處理。以下選項可用(默認為‘propagate’):
‘propagate’:返回 nan
‘raise’:引發錯誤
‘omit’:執行忽略 nan 值的計算
- alternative: {‘雙麵’,‘less’, ‘greater’},可選
定義備擇假設。默認為“雙麵”。可以使用以下選項:
“雙邊”:相關性不為零
‘less’:相關性為負(小於零)
‘greater’:相關性為正(大於零)
- res: SignificanceResult
包含屬性的對象:
- 統計 float 或 ndarray(二維正方形)
Spearman 相關矩陣或相關係數(如果僅給出 2 個變量作為參數)。相關矩陣是正方形,長度等於
a和b中變量(列或行)總數的總和。- p值 浮點數
假設檢驗的 p 值,其原假設是兩個樣本沒有序數相關性。有關替代假設,請參閱上麵的替代方案。 pvalue 與統計量具有相同的形狀。
ConstantInputWarning如果輸入是常量數組,則引發。這種情況下沒有定義相關係數,所以返回
np.nan。
參數 ::
返回 ::
警告 ::
參考:
[1]Zwillinger, D. 和 Kokoska, S. (2000)。 CRC 標準概率和統計表和公式。查普曼和霍爾:紐約。 2000. 第 14.7 條
[2]肯德爾,M.G. 和斯圖爾特,A. (1973)。高級統計理論,第 2 卷:推理和關係。格裏芬。 1973.第31.18條
[3]Kershenobich, D.、Fierro, F. J. 和 Rojkind, M. (1970)。人肝硬化中遊離脯氨酸池與膠原蛋白含量的關係。臨床研究雜誌,49(12), 2246-2249。
[4]Hollander, M.、Wolfe, D. A. 和 Chicken, E. (2013)。非參數統計方法。約翰·威利父子。
[5]B. Phipson 和 G. K. Smyth。 “排列 P 值不應該為零:隨機抽取排列時計算精確的 P 值。”遺傳學和分子生物學中的統計應用 9.1 (2010)。
[6]勒德布魯克,J. 和達德利,H. (1998)。為什麽排列檢驗在生物醫學研究中優於 t 和 F 檢驗。 《美國統計學家》,52(2), 127-132。
例子:
考慮以下來自[3]的數據,該數據研究了不健康人類肝髒中遊離脯氨酸(一種氨基酸)和總膠原蛋白(一種常見於結締組織中的蛋白質)之間的關係。
下麵的
x和y數組記錄了兩種化合物的測量值。觀察結果是配對的:每個遊離脯氨酸測量值均取自同一肝髒,作為相同index下的總膠原蛋白測量值。>>> import numpy as np >>> # total collagen (mg/g dry weight of liver) >>> x = np.array([7.1, 7.1, 7.2, 8.3, 9.4, 10.5, 11.4]) >>> # free proline (μ mole/g dry weight of liver) >>> y = np.array([2.8, 2.9, 2.8, 2.6, 3.5, 4.6, 5.0])在[4]中使用斯皮爾曼相關係數對這些數據進行了分析,這是一種對樣本之間單調相關性敏感的統計量。
>>> from scipy import stats >>> res = stats.spearmanr(x, y) >>> res.statistic 0.7000000000000001對於具有強正序相關的樣本,該統計量的值往往較高(接近 1);對於具有強負序相關的樣本,該統計量的值往往較低(接近 -1);對於具有強負序相關的樣本,該統計量的值往往較小(接近於 0)。具有弱序數相關性。
該檢驗是通過將統計觀察值與零分布進行比較來進行的:零假設下得出的統計值分布,即總膠原蛋白和遊離脯氨酸測量值是獨立的。
對於此測試,可以轉換統計量,使得大樣本的零分布是具有
len(x) - 2自由度的 Student t 分布。>>> import matplotlib.pyplot as plt >>> dof = len(x)-2 # len(x) == len(y) >>> dist = stats.t(df=dof) >>> t_vals = np.linspace(-5, 5, 100) >>> pdf = dist.pdf(t_vals) >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> def plot(ax): # we'll re-use this ... ax.plot(t_vals, pdf) ... ax.set_title("Spearman's Rho Test Null Distribution") ... ax.set_xlabel("statistic") ... ax.set_ylabel("probability density") >>> plot(ax) >>> plt.show()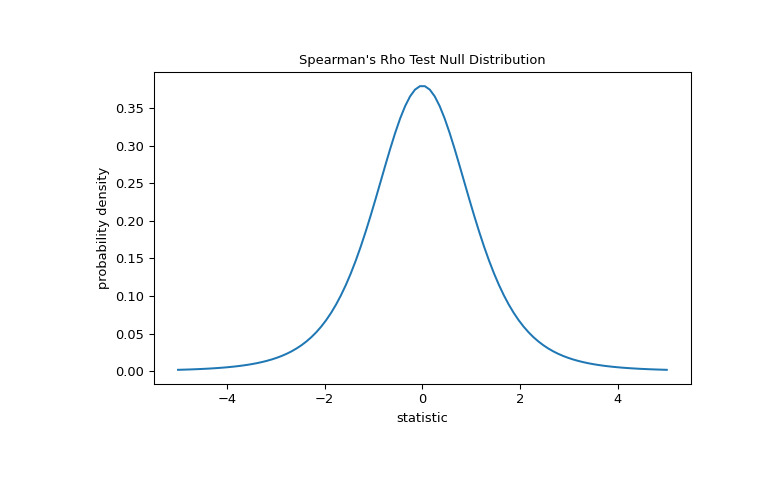
比較通過 p 值進行量化:零分布中比統計觀察值極端或更極端的值的比例。在統計量為正的雙邊測試中,大於轉換統計量的零分布元素和小於觀察統計量負值的零分布元素均被視為 “more extreme”。
>>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> rs = res.statistic # original statistic >>> transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs))) >>> pvalue = dist.cdf(-transformed) + dist.sf(transformed) >>> annotation = (f'p-value={pvalue:.4f}\n(shaded area)') >>> props = dict(facecolor='black', width=1, headwidth=5, headlength=8) >>> _ = ax.annotate(annotation, (2.7, 0.025), (3, 0.03), arrowprops=props) >>> i = t_vals >= transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> i = t_vals <= -transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> ax.set_xlim(-5, 5) >>> ax.set_ylim(0, 0.1) >>> plt.show()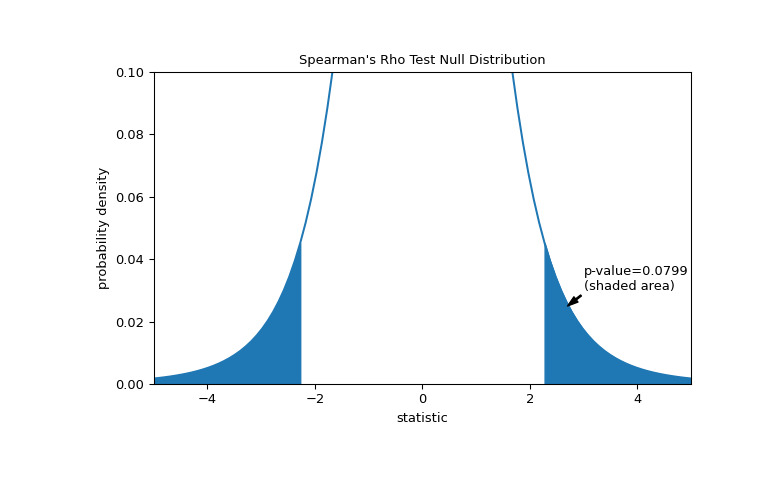
>>> res.pvalue 0.07991669030889909 # two-sided p-value如果 p 值為 “small” - 也就是說,如果從產生如此統計量極值的獨立分布中采樣數據的概率較低 - 這可以作為反對零假設的證據,支持替代方案:總膠原蛋白和遊離脯氨酸的分布不是獨立的。注意:
反之則不成立;也就是說,檢驗不用於為原假設提供證據。
將被視為 “small” 的值的閾值是在分析數據之前應做出的選擇 [5],同時考慮誤報(錯誤地拒絕原假設)和漏報(未能拒絕假設)的風險。錯誤的原假設)。
p 值小並不能證明效果大;相反,它們隻能為 “significant” 效應提供證據,這意味著它們不太可能在原假設下發生。
假設在進行實驗之前,作者有理由預測總膠原蛋白和遊離脯氨酸測量值之間存在正相關性,並且他們選擇評估零假設相對於片麵替代方案的合理性:遊離脯氨酸具有正相關性與總膠原蛋白的順序相關。在這種情況下,隻有零分布中與觀察到的統計量一樣大或更大的值才被認為是更極端的。
>>> res = stats.spearmanr(x, y, alternative='greater') >>> res.statistic 0.7000000000000001 # same statistic >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> pvalue = dist.sf(transformed) >>> annotation = (f'p-value={pvalue:.6f}\n(shaded area)') >>> props = dict(facecolor='black', width=1, headwidth=5, headlength=8) >>> _ = ax.annotate(annotation, (3, 0.018), (3.5, 0.03), arrowprops=props) >>> i = t_vals >= transformed >>> ax.fill_between(t_vals[i], y1=0, y2=pdf[i], color='C0') >>> ax.set_xlim(1, 5) >>> ax.set_ylim(0, 0.1) >>> plt.show()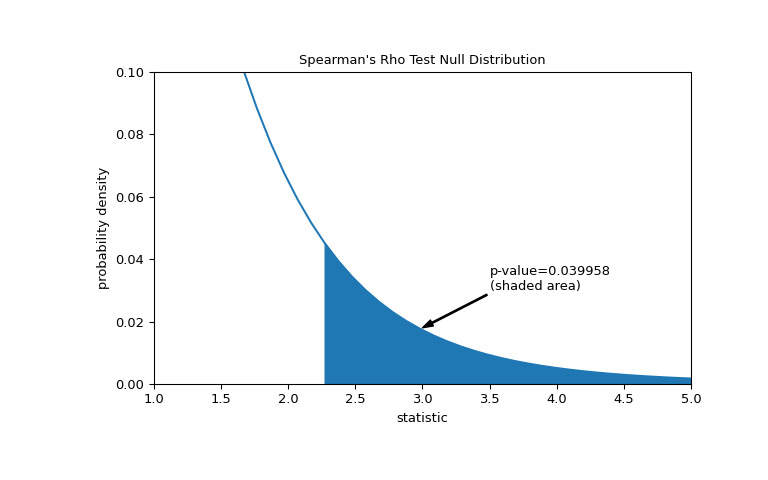
>>> res.pvalue 0.03995834515444954 # one-sided p-value; half of the two-sided p-value請注意,t 分布提供了零分布的漸近近似;它僅對於具有許多觀測值的樣本是準確的。對於小樣本,進行排列檢驗可能更合適:在總膠原蛋白和遊離脯氨酸獨立的零假設下,每個遊離脯氨酸測量值與任何總膠原蛋白測量值同樣可能被觀察到。因此,我們可以組建一個精確的通過計算之間每個可能的元素配對下的統計量來計算空分布
x和y.>>> def statistic(x): # explore all possible pairings by permuting `x` ... rs = stats.spearmanr(x, y).statistic # ignore pvalue ... transformed = rs * np.sqrt(dof / ((rs+1.0)*(1.0-rs))) ... return transformed >>> ref = stats.permutation_test((x,), statistic, alternative='greater', ... permutation_type='pairings') >>> fig, ax = plt.subplots(figsize=(8, 5)) >>> plot(ax) >>> ax.hist(ref.null_distribution, np.linspace(-5, 5, 26), ... density=True) >>> ax.legend(['aymptotic approximation\n(many observations)', ... f'exact \n({len(ref.null_distribution)} permutations)']) >>> plt.show()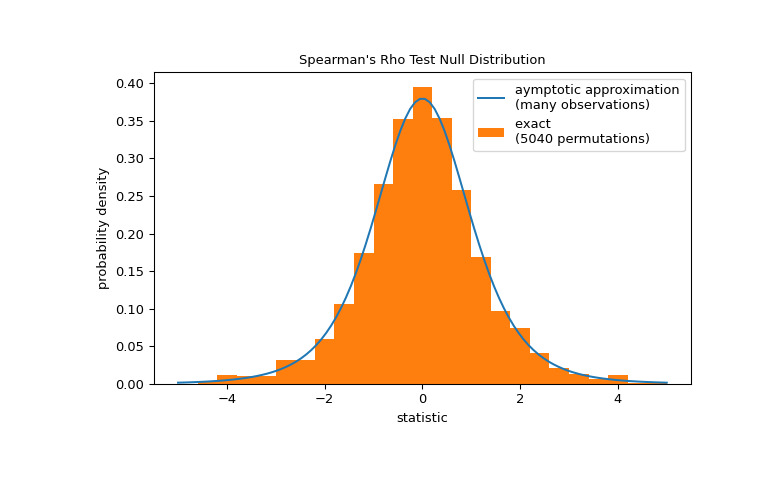
>>> ref.pvalue 0.04563492063492063 # exact one-sided p-value
相關用法
- Python SciPy stats.special_ortho_group用法及代碼示例
- Python SciPy stats.skewnorm用法及代碼示例
- Python SciPy stats.semicircular用法及代碼示例
- Python SciPy stats.skellam用法及代碼示例
- Python scipy.stats.somersd用法及代碼示例
- Python SciPy stats.sem用法及代碼示例
- Python SciPy stats.siegelslopes用法及代碼示例
- Python SciPy stats.skewcauchy用法及代碼示例
- Python SciPy stats.scoreatpercentile用法及代碼示例
- Python SciPy stats.skew用法及代碼示例
- Python SciPy stats.shapiro用法及代碼示例
- Python SciPy stats.studentized_range用法及代碼示例
- Python SciPy stats.sigmaclip用法及代碼示例
- Python SciPy stats.skewtest用法及代碼示例
- Python SciPy stats.sobol_indices用法及代碼示例
- Python SciPy stats.anderson用法及代碼示例
- Python SciPy stats.iqr用法及代碼示例
- Python SciPy stats.genpareto用法及代碼示例
- Python SciPy stats.cosine用法及代碼示例
- Python SciPy stats.norminvgauss用法及代碼示例
- Python SciPy stats.directional_stats用法及代碼示例
- Python SciPy stats.invwishart用法及代碼示例
- Python SciPy stats.bartlett用法及代碼示例
- Python SciPy stats.levy_stable用法及代碼示例
- Python SciPy stats.page_trend_test用法及代碼示例
注:本文由純淨天空篩選整理自scipy.org大神的英文原創作品 scipy.stats.spearmanr。非經特殊聲明,原始代碼版權歸原作者所有,本譯文未經允許或授權,請勿轉載或複製。